Human estrogen receptor alpha (ERα) targeted cyclic peptides inhibit cell growth and induce apoptosis in MCF-7 cells
- Emre Cetin
- 11 Eki 2024
- 21 dakikada okunur
Güncelleme tarihi: 17 Eki 2024
Abstract
Objectives
Human estrogen receptor alpha (ERα) is considered an important target, especially in the treatment of breast cancer, as it has a vital role in cancer development. ERα-targeted therapies generally target the ligand binding domain (LBD) of ERα. However, over time, cells develop resistance to this mechanism alternative approaches to inhibit ERα activity target ERα–DNA or ERα–cofactor interactions. Inhibitors of ERα–cofactor interactions are designed by targeting the hydrophobic hollow region of the receptor box LXXLL motif.
Methods
In this context, helix-stabilized cyclic peptides (SPs) designed with in silico approaches were obtained by solid phase peptide synthesis. The effects of SPs on MCF-7 cells were examined with MTT and ATP, and qPCR and flow cytometry were used for further analysis.
Results
Our results demonstrated that the SPs were effective only in MCF-7 cells expressing ERα. In addition, cyclic peptide combinations (SPCs) showed anti-proliferative and toxic effects on MCF-7 cells. The impact of SPCs with the highest inhibitory effect in MCF-7 cells on ERα-related genes and markers of apoptosis was revealed. Moreover, the flow cytometry analysis result used to examine apoptotic cells proved the apoptosis of SPCs in MCF-7 cells.
Conclusions
These findings suggest that our novel SPs, which inhibit coactivator interactions of ERα, induce apoptosis of MCF-7 cells. Thus, considering this strong effect of SPs in the inhibition of receptors, it is pointed out that they can be further developed as an alternative to current clinical treatments or as an auxiliary approach in the generating of new targeted peptide-based therapies.
Keywords: apoptosis; breast cancer; coactivator binding inhibitors; helix-stabilized cyclic peptides; human estrogen receptor alpha
Introduction
Human estrogen receptor alpha (ERα) is a ligand-dependent transcription factor [1, 2], and mediated gene products affect various cell processes such as autophagy, apoptosis, growth, and differentiation [3]. In addition to being associated with a variety of diseases, overexpressing estrogen receptor signaling contributes to disease progression [4], particularly in hormone-related cancers such as breast [5], endometrial [6], and ovarian cancer [7]. Because of the resistance that cancer cells develop over time to traditional ERα-targeted therapies, new alternative approaches have been developed [8, 9]. These approaches include targeting ERα–DNA or ERα–cofactor interactions (LXXLL) [10, 11]. Inhibiting protein–protein interactions (PPIs), such as ERα–cofactor interactions, with therapeutic peptides has become a general strategy in the treatment [12, 13]. Therapeutic cyclic peptides are preferred because they have various advantages, such as higher stability, easier production, and better cell permeability than their linear motifs [14, 15]. Various therapeutic cyclic peptides containing the LXXLL peptide motif [16], [17], [18], [19], [20], [21] have been developed to inhibit the ERα–coactivator interaction. Novel disulfide bridges stabilized cyclic peptide analogs are an effective treatment method for breast and ovarian cancer with high ERα expression. The aim of this study is to obtain disulfide-linked cyclic peptides that inhibit coactivator interactions of ERα and to elucidate their anti-proliferative and apoptosis effects on the MCF-7 breast cancer cells.
Materials and methods
Materials
This section is detailed in the Supplementary Material.
Molecular docking
The parent helix-stabilized cyclic peptides’ (SPs) crystal structure was retrieved from the Protein Data Bank (PDB ID: 1PCG). The sequence of this parent cyclic peptide in the crystal structure is KcILCRLLQ, where the lowercase ‘c’ represents the d-enantiomer of cysteine. Single mutations were introduced at the third position of the KcILCRLLQ sequence to generate child sequences, which were subject to docking using the bioAIM software from Meddenovo Drug Design. The best pose of each child sequence was retained, and its corresponding complexe formed with the receptor was prepared for molecular dynamic simulations (MD). Simulations were performed using the AMBER20 software [22]. Each complexe was solvated, neutralized, minimized and heated in a stepwise fashion (from 0 to 150 K in the constant temperature, constant volume (NVT) ensemble and from 150 to 300 K in the constant temperature, constant pressure (NPT) ensemble). In all simulations, periodic boundary conditions were applied with the particle mesh Ewald scheme for electrostatic interactions, the non-bonded interaction cut-off was set to 12.0 Å, bonds involving hydrogen atoms were constrained using the SHAKE algorithm, and the integration time step was set to 2 fs. Production runs were performed in the NPT ensemble for 5 ns, from which 5 independent 1 ns-long MD simulations were started and used for binding energy calculations. The average binding energy was calculated by applying molecular mechanics with generalized Born (igb=8) and surface area solvation (MMGBSA) to these simulations [23].
Synthesis, characterization, and purification of SPs
As a result of our in silico studies, the synthesis of a total of three SPs, including the recommended sequences and the reference sequence, was carried out by the solid-phase peptide synthesis method based on the fluorenyl methoxycarbonyl protecting group (Fmoc) chemistry. The synthesis of peptides was carried out as previously mentioned [24]. The crude linear peptide was dissolved in dimethylformamide (DMF)/H2O (68-12-2, Meck) 8:2 (10 mg/mL) and stirred vigorously at room temperature to get cyclic peptide. A solution of diethylacetylene dicarboxylate (DEAD) (1972-28-7, Merck) in DMF (1 eq./mL) was slowly added over 2 min, and the reaction was left stirring for 30 min. The resulting mixture was diluted to 20 mL with MilliQ water, and a freeze dryer removed the solvent. Obtained cyclic peptides were purified using a semi-preparative C18 high-performance liquid chromatography (HPLC) column. Analytical C18 HPLC evaluated the purity of each fraction before combining the pure fractions. In all HPLC analyses, the mobile phase was gradient elution, MilliQ water – 0.1 % trifluoroacetic acid (TFA) (T6508, Meck), and acetonitrile (ACN) (75-05-8, Meck) – 0.08 % TFA. The characterization was performed by liquid chromatography-mass spectrometry (LC-MS).
Cell culture
Human breast cancer (BC) cell lines (ERα-positive MCF-7 and ERα-negative MDA-MB-231) and human bronchial epithelial cell line (BEAS-2B) were cultured in RPMI 1640 (R8758, Sigma) containing 1 % penicillin streptomycin (PS-B, Capricorn), 1 % L-Glutamine (25030-081, Gibco), 10 % heat-inactivated fetal bovine serum (10100147, Thermo Fisher) and β-estradiol (E2758, Sigma). All cell lines were maintained at 37 °C in an environment containing 5 % CO2 and 98 % humidity. All SPs have >95 % purity and are lyophilized as TFA salts. 25 mM stock solutions of all SPs were prepared by dissolving in molecular grade water, while a stock solution of TPBM was prepared by DMSO (A3672.0100, Pan-reac).
Cell viability assays
The viability effects of SPs on MCF-7 cells using the MTT and ATP assays. For each experimental viability assay, MCF-7 cells were seeded in 96-well cell culture dishes at 5×103 cells/well, and to ensure the adaptation, the cells were incubated for 24 h. For MTT analysis, MCF-7 cells were treated with serial dilutions of all SPs and TPBM for 48 h. Following the treatment, an MTT assay was performed as described previously [25]. The level of intracellular ATP content is an indicator used to determine the number of living cells [26, 27]. This method was used in our study to examine the effect of combined treatment groups designed with all SPs and TPBM on cell viability. Following the treatment, an ATP assay was performed as described previously [27]. Each experiment was carried out as three independent tests with three replicated.
Gene expression analysis
Total ribonucleic acid (RNA) was isolated with GenElute™ mammalian total RNA miniprep kit, and complementary deoxyribonucleic acid (cDNA) synthesis was conducted using SensiFAST cDNA synthesis kit according to the manufacturer’s instructions. The amount and purity of all RNA samples were measured by NanoDrop™ 2000/2000c spectrophotometer (thermo fisher scientific). Annealing temperatures of the primers used with gradient polymerase chain reaction (PCR) were determined. All PCR products were checked for product length with 2 % agarose gel. Real-time quantitative reverse transcription PCR (RT-qPCR) analysis was performed using a CFX96™ real-time PCR detection system and carried out as described previously [28]. Melting curve analysis was performed. Data were analyzed with stepone plus v2.3 (Applied Biosystems) software using the 2−ΔΔCT method.
Flow cytometry
To evaluate the effect of SPs on apoptosis, we measured caspase 3/7 activity, mitochondrial membrane potential, and annexin-V-FITC level by flow cytometry. Cells were seeded at 3×105 cells per well into 6-well plates and treated with SPCs and TPBM (IC90 doses) for 48 h. At the end of treatment, cells were harvested and prepared according to the manufacturer’s instructions for each kit, and analysis was performed by a cell analyzer [29].
Statistical analysis
Data were presented as mean±standard error of the mean (SEM). Analysis of variance was used to compare the control and treated groups. Student’s t-test, one-way ANOVA, and two-way ANOVA were used to determine p-values. p-Value less than 0.05 was considered statistically significant and p<0.05, * p<0.01, *** p<0.001, **** p<0.0001 indicated statistically significant results. Statistical analyses were performed using prism software (version 9.0.2; GraphPad software).
Results
In silico designing of potential SPs targeting the LXXLL motif of ERα
Visual analysis determined that replacing the isoleucine at the third position of the parent sequence with one of the positively charged or hydrophilic groups would increase the interaction. In addition, it was predicted that introducing a negatively charged amino acid would reduce the interaction and, therefore, the binding of the peptide. Protein–peptide complexes were formed by applying point mutations to the isoleucine amino acid in the crystal structure, and molecular dynamics simulations of the complexes were run to calculate the binding energy, as detailed in the Methods section. Binding affinities of the KcXLCRLLQ sequence (where X=I, S, H, K, D) against ERα are shown in (Table 1). According to binding energy calculations, hydrophilic (S) and positively charged (K) amino acids increased the binding affinity compared to the reference (I). In the direction of these results, two cyclic peptides with high ERα-specific affinity SP1: KcILCRLLQ (as a reference peptide), SP2: Kc*KLCRLLQ and SP3: Kc*SLCRLLQ were selected for in vitro experiments (Table 2). 3D structure of ERα with complexed docked SP1 (Supplementary Figure 1A), SP2 (Supplementary Figure 2A), and SP3 (Supplementary Figure 3A) and the amino acid residues of candidate SPs were visualized using LigPlot with a 2D display. The visualization results of the interaction between amino acid residues of SP1 (Supplementary Figure 1B), SP2 (Supplementary Figure 2B), and SP3 (Supplementary Figure 3B) and ERα highlighted bond and hydrophobic contacts.
Table 1:
Average MMGBSA binding energy calculations of MD simulations for ERα in complex with the SPs containing different amino acid residues in the X group for KCXLCRLLQ sequence.
Amino acid | Ile (I) | Ser (S) | His (H) | Lys (K) | Asp (D) |
Binding energy, kcal/mol | −42.7±3.4 | −44.7±4.0 | −36.8±3.7 | −42.9±3.5 | −36.7±3.6 |
ERα, human estrogen receptor alpha; MD, molecular dynamics; MMGBSA, generalised born and surface area solvation; SPs, helix-stabilized cyclic peptides; Ile, isoleucine; Ser, serine; His, histidine; Lys, lysine; Asp, aspartc acid.
Table 2:
Sequences of SPs in one letter amino acid code. The disulfide-linked cyclization forming between cysteine residues is formed between “c” and “C”.
Peptide | Sequence |
SP1 | K c I L C R L L Q |
SP2 | K c K L C R L L Q |
SP3 | K c S L C R L L Q |
SP1, helix-stabilized cyclic peptide 1; SP2, helix-stabilized cyclic peptide 2; SP3, helix-stabilized cyclic peptide 3.
Synthesis and characterization of SPs
Novel SPs (SP2: KcKLCRLLQ and SP3: KcSLCRLLQ), the reference sequence in the literature (SP1: KcILCRLLQ), and their linear peptides (LPs) were characterized by LC-MS. There are two major molecular ion peaks for both SP1 and LP1 in their mass spectrum (Supplementary Figure 4A and B). These are [M+H]+ and [M+2H]2+ for both peptides. The mass difference between [M+H]+ peaks for LP1 and SP1 is 2 Da, which shows the disulfide bridge formation due to the elimination of two hydrogen atoms in the cyclization reaction. There are three major molecular ion peaks in the mass spectrum of LP2 and SP2 (Supplementary Figure 5A and B). These are [M+H]+, [M+2H]2+ and [M+3H]3+ for both peptides. The mass difference between [M+H]+ peaks for LP2 and SP2 is 2 Da which showed the disulfide bridge formation due to the elimination of two hydrogen atoms in the cyclisation reaction. There are three major molecular ion peaks for both SP3 and LP3 in their mass spectrum (Supplementary Figure 6A and B). These are [M+H]+, [M+2H]2+ and [M+3H]3+ for both peptides. The mass difference between [M+H]+ peaks for LP3 and SP3 is 2 Da which showed the disulfide bridge formation due to the elimination of two hydrogen atoms in the cyclisation reaction.
Effect of SPs on ERα-positive cells in the presence and absence of estradiol (E2)
The effect of SP1(reference) and the novel SPs (SP2-3) on cell viability in the presence and absence of E2 was examined. For this, ERα-positive MCF-7 cells were treated different concentrations of SPs and TPBM (at the range of 50–6.25 μM) were treated in the presence (1 pM) and absence of E2 for 48 h. Cell viability at the end of treatment was measured by MTT assay. The degree of significance was measured by comparing each peptide with its control group. SPs and TPBM treated to cells in an E2-free medium did not show a statistically significant change in cell viability (Figure 1A), while in the presence of 1 pM, E2 significantly reduced cell proliferation at concentrations of 50 μM (Figure 1B). 50 μM of SPs and TPBM reduced cell viability by 37 % (p=0.0051), 27 % (p=0.0323), 23 % (p=0.0164), and 37 % (p=0.0048), respectively.

Figure 1:
Effect of SPs on MCF-7 cells cultured in different culture media. (A) Cell viability effects of SPs and TPBM on MCF-7 cells in the absence and (B) presence of E2. Bars represent the average±standard error of the mean (SEM). For statistical analysis of the MTT data, two-way ANOVA was performed.
Effect of SPs on the viability of cell lines with different ERα expression levels
To measure the effects of SPs on ERα (+) MCF-7 and ERα (−) MDA-MB-231 and BEAS-2B cells were treated with SPs and TPBM at different concentrations (100–6.25 μm) and incubated for 48 h. Results indicated that all cyclic peptides were not inhibitory on cell growth in ERα (−) cell lines but showed anti-proliferative effects only in ERα (+) MCF-7 cells. 50 and 100 μM concentrations of all cyclic peptides showed a significant decrease in cell viability on MCF-7 cells relative to control. SP1 reduced cell viability by 36.5 % (p=0.0038) and 41.4 % (p<0.0061) (Figure 2A). SP2 inhibited cell viability by 27.7 % (p=0.0407) and 46.65 % (p=0.0002), respectively (Figure 2B). SP3 reduced cell viability by 23.4 % (p=0.0172) and 43.15 % (p<0.001), respectively (Figure 2C). TPBM, showed an effect only on MCF-7 cells, like SPs. 50 and 100 μM of TPBM reduced cell viability compared to the control group by 39.7 % (p=0.0009) and 65.65 % (p<0.0001), respectively. From the MTT result, the half-maximal inhibitory concentration (IC50) value of TPBM was 63.3 μM (Figure 2D).

Figure 2:
The effect of SPs and TPBM on the cell viability of MDA-MB-231, BEAS-2B, and MCF-7 cells. For MTT results, bars represent the average±standard error of the mean (SEM). Two-way ANOVA was performed to analyze the MTT data statistically. p<0.05, *p<0.01,***p<0.001, ****p<0.0001.
Effect of SPs combinations and single SPs treatments on MCF-7 growth inhibition
ATP assay was used to examine the effect of combinations of TPBM and SPs on MCF-7 cells. For the first combination experiment, cells were pre-treated with an IC50 dose of TPBM. The next day, cells were treated with 50 μM of each SP for 48 h. For the second combination experiment, cells were pretreated with SPs and TPBM (12.5–100 μM of each). After 24 h, combination groups were generated by adding peptides to the group to which they were not applied. Each peptide was used at the same concentration (12.5–100 μM). According to the results, there was a significant decrease in cell viability of MCF-7 cells pre-treated with IC50 dose after treatment with SP1, SP2, and SP3 (Figure 3A). The TPBM+SP1+SP2+SP3 combination showed a toxic effect on the MCF-7 cells. For SPCs, it was observed that a potent anti-proliferative effect was in a dose-dependent manner in all groups (Figure 3B). Although SP1+SP2 and SP1+SP3 combinations also reduce cell number, the most effective combination treatment groups are SP2+SP3 and SP1+SP2+SP3. Our study’s only triple treatment group, SP1+SP2+SP3, showed the best anti-proliferative effect. At the same time, doses of 12.5, 25 and 50 µM reduced cell viability by 20.4 % (p<0.05), 65.1 % (p<0.0001) and 93.8 % (p<0.0001), respectively and the 100 µM dose was observed to have a toxic effect on the cells. While the IC50 value of SP1+SP2+SP3 was ∼21 µM, the IC90 value was ∼47 µM. As a result of ATP analysis in which the concentration of each peptide was increased for other peptide combinations, the IC90 value of SP2+SP3 was found to be ∼134 µM.

Figure 3:
The effect of TPBM and SPs combinations on the cell viability of MCF-7 cells. (A) viability rate of SPs treated cells after pre-treatment with IC50 dose of TPBM. (B) Viability rate of cells treated with the same doses after pre-treatment with different SPs concentrations. Bars represent the average±standard error of the mean (SEM). For statistical analysis of the ATP data, two-way ANOVA was performed. p<0.05, *p<0.01,***p<0.001, ****p<0.0001.
Effect of SPCs on the expression of genes associated with the apoptosis and ERα-related
MCF-7 cells were treated with IC50 doses of SPCs and TPBM for 48 h, and then the expression levels of ERα and its target genes and apoptosis markers (Supplementary Table 1). mRNA expressions were evaluated via qRT-PCR. Results indicated that SPCs and TPBM were effective in reducing mRNA expression of ESR1 and its target mRNA expressions (Figure 4A). All SPCs decreased the ESR1 mRNA expression level, and the most effective SPC was SP1+SP2+SP3, which reduced the expression level by ∼1.5-fold. The mRNA expression level of TFF1 decreased significantly more than 1.5 fold in all treatment groups (p<0.0001). The most effective combination treatments in down-regulating other ERα-related genes were SP1+SP3, SP2+SP3, and SP1+SP2+SP3 (Figure 4A). In addition, there has been a significant increase in the number of regulators of apoptosis in peptide combination groups. A substantial increase in Bax mRNA expression levels was observed in the SP1+SP2+SP3 group (p<0.05) (Figure 4B). There was a drop in Bcl-2 mRNA expression in all treatment groups, but in Bax:Bcl-2 ratios showed that in SP1+SP3, SP2+SP3, and SP1+SP2+SP3 increased significantly (p<0.0001). When caspase-8 and p21 mRNA expressions were compared to control, the most effective peptide combinations were SP2+SP3 and SP1+SP2+SP3. In particular, high p53 mRNA expression induces apoptosis in MCF-7 cells, and when comparing treatment groups, it was significantly increased in SP1+SP3 (p<0.05), SP2+SP3 (p<0.05), and SP1+SP2+SP3 (p<0.01) (Figure 4B).
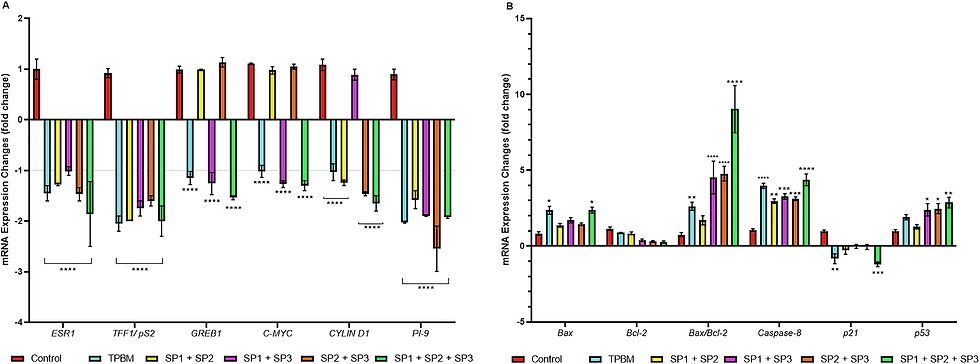
Figure 4:
RT-qPCR analysis of ERα and apoptosis-related mRNAs after being treated with IC50 of SPCs and TPBM on MCF-7 cells. Beta actin was used as a housekeeping gene. (A) ERα-mediated mRNA expression levels; (B) mRNA levels of apoptosis regulators after treatment. Bars represent the average±standard error of the mean (SEM). For statistical analysis of the qPCR data, two-way ANOVA was performed. p<0.05, *p<0.01,***p<0.001, ****p<0.0001.
Confirmation that SPCs stimulate apoptosis in MCF-7 cells by flow cytometry analysis
Following the evaluation of the apoptotic cell death mechanism with annexin-V in flow cytometry, the total apoptotic cell rates after 48 h of treatment with IC90 of SPCs in the MCF-7 cell line were 98.66 % (early+late) in the SP2+SP3 group, in the SP1+SP2+SP3 group, it was determined that the total apoptotic cells were 97.07 % (Figure 5A). As a result of the evaluation of caspase-3/7 analysis, the total apoptotic cell numbers after 48 h of SPC treatment in the MCF-7 cell line were measured as 99.6 % (early+late) in the SP2+SP3 group, while in the SP1+SP2+SP3 group was determined as 96.75 % (Figure 5B). While late apoptotic changes increased with triple peptide treatment, both early and late apoptotic changes differed between SPCs. Since MCF-7 cells do not have caspase-3 expression [30], only the increase in the assay is caspase seven activation. All these results showed that the treatment of SPCs significantly increased the caspase-3/7 activity in the MCF-7 cells. While the rate of total cells in which the mitochondrial membrane potential changed was 25.65 % in the SP2+SP3 group, this rate was found to be 53.55 % in the SP1+SP2+SP3 group (Figure 5C).
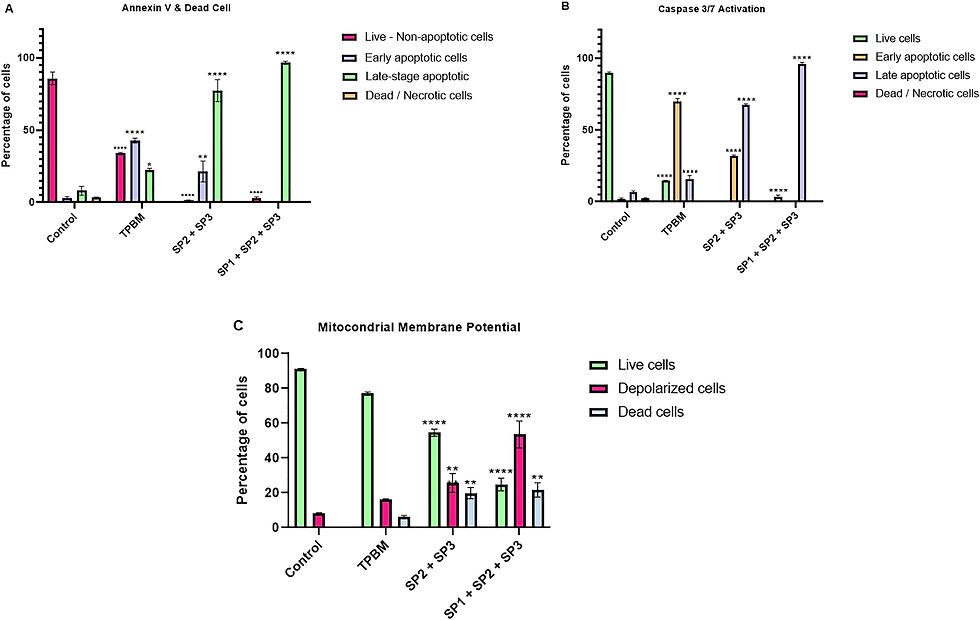
Figure 5:
Assessment of (A) annexin V-FITC, (B) caspase-3/7, and (C) mitochondrial membrane potential change activation on MCF-7 cells were exposed to IC90 values of SPCs and TPBM for 48 h. (A and B) the percentages of live, dead, early apoptotic, and late apoptotic cells are presented in each graph. (C) The effects on mitochondrial membrane potential were shown as a percentage, dividing the cells into live dead and total depolarized cells. The percentage of cells is represented as the average±standard error of the mean (SEM) from at least two independent experiments. Two-way ANOVA was performed to analyze the flow cytometry data statistically. p<0.05, *p<0.01,***p<0.001, ****p<0.0001.
Discussion
Peptides with the LXXLL motif have been used to design peptide mimics that bind to the surface of steroid receptors and may be offered as candidate treatments in some cases of resistance to therapeutics currently used in the clinic [31], [32], [33]. Disulfide bonds stabilize linear peptides because they play an essential role in the biological effects of many peptide drugs [34, 35]. A few studies in the literature of disulfide bridged peptides developed specifically targeted the co-activator region of ERα. The first disulfide bond-stabilized cyclic (PERM-1) that inhibits the ERα-cofactor binding site was created by Leduc et al. [18]. In subsequent studies, different analogs of PERM-1 were designed. These were shown to have a higher helical character and a lower inhibition constant against ERα [36], showed a higher affinity to ERα [16], and could downregulate the mRNA expression of the relevant gene pS2 (TFF1) by significantly affecting ERα-mediated transcription [37, 38]. In this study, new therapeutic ERα-targeted cyclic peptides with disulfide bridges and tested the effectiveness of our therapeutic peptide candidates on MCF-7 cells. TPBM commercial small molecule was used as a reference inhibitor in our study because it does not bind to the ligand binding domain (LBD) region of estrogen receptor alpha without competing with estrogen [39] and causes a function similar to the mechanism of action of our peptides. The SPs developed in our research are specifically designed for ERα in its active form (complexed with E2). Supporting this design model, our in vitro results showed that SPs in E2-deprived medium did not have any effect on MCF-7 cells, and the anti-proliferative effects of SPs were observed only in the presence of the optimum E2 concentration. At the same time, SPs in an E2-supplemented cell culture medium did not show any toxic effect on cancer and normal cells that do not express ERα. It showed that the peptides inhibited cell growth via ERα. However, since the IC50 value of single treatment of SPs up to 100 µM on MCF-7 cells could not be achieved, it was decided that treat to MCF-7 cells with SPCs. The results clearly demonstrated the growth inhibition of SPCs in MCF-7 cells. While SPs applied to MCF-7 cells pre-treated with TPBM effectively further reduced viability, significant anti-proliferative and toxic effects were also observed in groups treated with SPCs alone. In qPCR results confirmed that mRNA expression levels of ERα and its associated intracellular targets in MCF-7 cells treated with IC50 doses of SPCs were significantly down-regulated, thus inhibiting ERα and its targets. It was emphasized that the mRNA expression profile of the important apoptosis markers significantly induced apoptosis in MCF-7 cells after SPCs treatment. Additionally, flow cytometry was used to better analyze the apoptosis process, and it was determined that the number of annexin-V positive cells increased in MCF-7 cells treated with the IC90 dose of SPCs. Similarly, caspase-3/7 activities caused a decrease in cell viability and an increase in the percentage of apoptotic cells, and increases in the number of depolarized cells proved the apoptosis-increasing effect of SPCs in MCF-7 cells. As a result of our study, the fact that these new ERα-directed SPs showed inhibitory activity only against ERα (+) MCF-7 cells and stimulated apoptosis showed that SPs could be an alternative to existing clinical treatments or a candidate approach for the creation of new peptide combination therapies.
Corresponding author: Hilal Şentürk, Department of Biotechnology, Institute of Health Sciences, Bezmialem Vakif University, Fatih, Istanbul, Türkiye, E-mail: sentr.hilal@gmail.com
Award Identifier / Grant number: 223S648
Funding source: Health Institutes of Türkiye (TUSEB)
Award Identifier / Grant number: 12162
Acknowledgments
The authors thank TUBITAK and TUSEB for their support. Also, we would like to thank Murat Kaya Köşger for his contributions to the data analysis.
Research ethics: Not applicable.
Informed consent: Not applicable.
Author contributions: HŞ and HD designed the study. HŞ, IUM, SÖ, MSK, ŞÜB, MA, and MEG participated in generating the data for the study. HŞ, HD, IUM, SÖ, MSK, ŞÜB, MA and MEG participated in the analysis of the data. HŞ wrote the majority of the original draft of the paper. HŞ, HD, IUM, SÖ, MSK, ŞÜB, MA, MEG, FA and EU participated in writing the paper. The authors have accepted responsibility for the entire content of this manuscript and approved its submission.
Competing interests: The authors state no conflict of interest.
Research funding: This study was supported by the Scientific and Technological Research Council of Turkey (TUBITAK) under Grant Number 223S648 and the Health Institutes of Turkey (TUSEB) under Grant Number 12162.
Data availability: The data that support the findings of this study will be made available upon reasonable request.
References
1. Candelaria, NR, Liu, K, Lin, CY. Estrogen receptor alpha: molecular mechanisms and emerging insights. J Cell Biochem 2013;114:2203–8. https://doi.org/10.1002/jcb.24584.Search in Google ScholarPubMed
2. Welboren, WJ, Sweep, FC, Span, PN, Stunnenberg, HG. Genomic actions of estrogen receptor alpha: what are the targets and how are they regulated? Endocr Relat Cancer 2009;16:1073–89. https://doi.org/10.1677/erc-09-0086.Search in Google Scholar
3. Chen, P, Li, B, Ou-Yang, L. Role of estrogen receptors in health and disease. Front Endocrinol 2022;13:2–9. https://doi.org/10.3389/fendo.2022.839005.Search in Google ScholarPubMed PubMed Central
4. Faltas, CL, LeBron, KA, Holz, MK. Unconventional estrogen signaling in health and disease. Endocrinology 2020;161:3–14. https://doi.org/10.1210/endocr/bqaa030.Search in Google ScholarPubMed PubMed Central
5. Ali, S, Coombes, RC. Estrogen receptor alpha in human breast cancer: occurrence and significance. J Mammary Gland Biol Neoplasia 2000;5:271–81. https://doi.org/10.1023/a:1009594727358.10.1023/A:1009594727358Search in Google ScholarPubMed
6. Rodriguez, AC, Blanchard, Z, Maurer, KA, Gertz, J. Estrogen signaling in endometrial cancer: a key oncogenic pathway with several open questions. Horm Cancer 2019;10:51–63. https://doi.org/10.1007/s12672-019-0358-9.Search in Google ScholarPubMed PubMed Central
7. Chan, KKL, Siu, MKY, Jiang, YX, Wang, JJ, Leung, THY, Ngan, HYS. Estrogen receptor modulators genistein, daidzein and ERB-041 inhibit cell migration, invasion, proliferation and sphere formation via modulation of FAK and PI3K/AKT signaling in ovarian cancer. Cancer Cell Int 2018;18:65. https://doi.org/10.1186/s12935-018-0559-2.Search in Google ScholarPubMed PubMed Central
8. Criscitiello, C, Fumagalli, D, Saini, KS, Loi, S. Tamoxifen in early-stage estrogen receptor-positive breast cancer: overview of clinical use and molecular biomarkers for patient selection. OncoTargets Ther 2010;4:1–11. https://doi.org/10.2147/ott.s10155.Search in Google ScholarPubMed PubMed Central
9. Orlando, L, Schiavone, P, Fedele, P, Calvani, N, Nacci, A, Rizzo, P, et al.. Molecularly targeted endocrine therapies for breast cancer. Cancer Treat Rev 2010;36:S67–71. https://doi.org/10.1016/s0305-7372(10)70023-2.Search in Google ScholarPubMed
10. Musgrove, EA, Sutherland, RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer 2009;9:631–43. https://doi.org/10.1038/nrc2713.Search in Google ScholarPubMed
11. Shanle, EK, Xu, W. Selectively targeting estrogen receptors for cancer treatment. Adv Drug Deliv Rev 2010;62:1265–76. https://doi.org/10.1016/j.addr.2010.08.001.Search in Google ScholarPubMed PubMed Central
12. Bruzzoni-Giovanelli, H, Alezra, V, Wolff, N, Dong, CZ, Tuffery, P, Rebollo, A. Interfering peptides targeting protein–protein interactions: the next generation of drugs? Drug Discov Today 2018;23:272–85. https://doi.org/10.1016/j.drudis.2017.10.016.Search in Google ScholarPubMed
13. London, N, Raveh, B, Movshovitz-Attias, D, Schueler-Furman, O. Can self-inhibitory peptides be derived from the interfaces of globular protein–protein interactions? Proteins 2010;78:3140–9. https://doi.org/10.1002/prot.22785.Search in Google ScholarPubMed PubMed Central
14. Joo, SH. Cyclic peptides as therapeutic agents and biochemical tools. Biomol Ther 2012;20:19–26. https://doi.org/10.4062/biomolther.2012.20.1.019.Search in Google ScholarPubMed PubMed Central
15. Sanner, MF, Zoghebi, K, Hanna, S, Mozaffari, S, Rahighi, S, Tiwari, RK, et al.. Cyclic peptides as protein kinase inhibitors: structure–activity relationship and molecular modeling. J Chem Inf Model 2021;61:3015–26. https://doi.org/10.1021/acs.jcim.1c00320.Search in Google ScholarPubMed PubMed Central
16. Galande, AK, Bramlett, KS, Trent, JO, Burris, TP, Wittliff, JL, Spatola, AF. Potent inhibitors of LXXLL-based protein–protein interactions. Chembiochem 2005;6:1991–8. https://doi.org/10.1002/cbic.200500083.Search in Google ScholarPubMed
17. Geistlinger, TR, McReynolds, AC, Guy, RK. Ligand-selective inhibition of the interaction of steroid receptor coactivators and estrogen receptor isoforms. Chem Biol 2004;11:273–81. https://doi.org/10.1016/s1074-5521(04)00013-4.Search in Google Scholar
18. Leduc, AM, Trent, JO, Wittliff, JL, Bramlett, KS, Briggs, SL, Chirgadze, NY, et al.. Helix-stabilized cyclic peptides as selective inhibitors of steroid receptor–coactivator interactions. Proc Natl Acad Sci U S A 2003;100:11273–8. https://doi.org/10.1073/pnas.1934759100.Search in Google ScholarPubMed PubMed Central
19. Phillips, C, Roberts, LR, Schade, M, Bazin, R, Bent, A, Davies, NL, et al.. Design and structure of stapled peptides binding to estrogen receptors. J Am Chem Soc 2011;133:9696–9. https://doi.org/10.1021/ja202946k.Search in Google ScholarPubMed
20. Speltz, TE, Fanning, SW, Mayne, CG, Fowler, C, Tajkhorshid, E, Greene, GL, et al.. Stapled peptides with γ-methylated hydrocarbon chains for the estrogen receptor/coactivator interaction. Angew Chem Int Ed Engl 2016;55:4252–5. https://doi.org/10.1002/anie.201510557.Search in Google ScholarPubMed PubMed Central
21. Geistlinger, TR, Guy, RK. Novel selective inhibitors of the interaction of individual nuclear hormone receptors with a mutually shared steroid receptor coactivator 2. J Am Chem Soc 2003;125:6852–3. https://doi.org/10.1021/ja0348391.Search in Google ScholarPubMed
22. Macke, TJ, Svrcek-Seiler, W, Brown, RA, Kolossváry, I, Bomble, YJ, Case, DA, et al.. AmberTools users’ manual. Ver. Utah, USA: ACS; 2010.Search in Google Scholar
23. Genheden, S, Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov 2015;10:449–61. https://doi.org/10.1517/17460441.2015.1032936.Search in Google ScholarPubMed PubMed Central
24. Nezir, AE, Khalily, MP, Gulyuz, S, Ozcubukcu, S, Küçükgüzel, ŞG, Yilmaz, O, et al.. Synthesis and evaluation of tumor-homing peptides for targeting prostate cancer. Amino Acids 2021;53:645–52. https://doi.org/10.1007/s00726-021-02971-3.Search in Google ScholarPubMed
25. Ulukaya, E, Ari, F, Dimas, K, Ikitimur, EI, Guney, E, Yilmaz, VT. Anti-cancer activity of a novel palladium (II) complex on human breast cancer cells in vitro and in vivo. Eur J Med Chem 2011;46:4957–63. https://doi.org/10.1016/j.ejmech.2011.07.055.Search in Google ScholarPubMed
26. Andreotti, PE, Cree, IA, Kurbacher, CM, Hartmann, DM, Linder, D, Harel, G, et al.. Chemosensitivity testing of human tumors using a microplate adenosine triphosphate luminescence assay: clinical correlation for cisplatin resistance of ovarian carcinoma. Cancer Res 1995;55:5276–82.Search in Google Scholar
27. Ulukaya, E, Ozdikicioglu, F, Oral, AY, Demirci, M. The MTT assay yields a relatively lower result of growth inhibition than the ATP assay depending on the chemotherapeutic drugs tested. Toxicol Vitro 2008;22:232–9. https://doi.org/10.1016/j.tiv.2007.08.006.Search in Google ScholarPubMed
28. Bender, O, Gunduz, M, Cigdem, S, Hatipoglu, OF, Acar, M, Kaya, M, et al.. Functional analysis of ESM 1 by si RNA knockdown in primary and metastatic head and neck cancer cells. J Oral Pathol Med 2018;47:40–7. https://doi.org/10.1111/jop.12648.Search in Google ScholarPubMed
29. Akgun, O, Erkisa, M, Ari, F. Effective and new potent drug combination: histone deacetylase and Wnt/β-catenin pathway inhibitors in lung carcinoma cells. J Cell Biochem 2019;120:15467–82. https://doi.org/10.1002/jcb.28813.Search in Google ScholarPubMed
30. Liang, Y, Yan, C, Schor, NF. Apoptosis in the absence of caspase 3. Oncogene 2001;20:6570–8. https://doi.org/10.1038/sj.onc.1204815.Search in Google ScholarPubMed
31. Qin, W, Xie, M, Qin, X, Fang, Q, Yin, F, Li, Z. Recent advances in peptidomimetics antagonists targeting estrogen receptor α-coactivator interaction in cancer therapy. Bioorg Med Chem Lett 2018;28:2827–36. https://doi.org/10.1016/j.bmcl.2018.05.062.Search in Google ScholarPubMed
32. Verdine, GL, Hilinski, GJ. Stapled peptides for intracellular drug targets. Methods Enzymol 2012;503:3–33. https://doi.org/10.1016/b978-0-12-396962-0.00001-x.Search in Google ScholarPubMed
33. Skowron, KJ, Booker, K, Cheng, C, Creed, S, David, BP, Lazzara, PR, et al.. Steroid receptor/coactivator binding inhibitors: an update. Mol Cell Endocrinol 2019;493:110471.10.1016/j.mce.2019.110471Search in Google ScholarPubMed PubMed Central
34. Schteingart, CD, Lau, JL. From natural peptides to market. In: Goodnow, RAJr, editor. Annual reports in medicinal chemistry. Boston, MA, United States: Elsevier; 2017.10.1016/bs.armc.2017.08.003Search in Google Scholar
35. Johnson, M, Liu, M, Struble, E, Hettiarachchi, K. Characterization of cyclic peptides containing disulfide bonds. J Pharm Biomed Anal 2015;109:112–20. https://doi.org/10.1016/j.jpba.2015.01.009.Search in Google ScholarPubMed PubMed Central
36. Galande, AK, Bramlett, KS, Burris, TP, Wittliff, JL, Spatola, AF. Thioether side chain cyclization for helical peptide formation: inhibitors of estrogen receptor–coactivator interactions. J Pept Res 2004;63:297–302. https://doi.org/10.1111/j.1399-3011.2004.00152.x.Search in Google ScholarPubMed
37. Nagakubo, T, Demizu, Y, Kanda, Y, Misawa, T, Shoda, T, Okuhira, K, et al.. Development of cell-penetrating R7 fragment-conjugated helical peptides as inhibitors of estrogen receptor-mediated transcription. Bioconjug Chem 2014;25:1921–4. https://doi.org/10.1021/bc500480e.Search in Google ScholarPubMed
38. Demizu, Y, Misawa, T, Nagakubo, T, Kanda, Y, Okuhira, K, Sekino, Y, et al.. Structural development of stabilized helical peptides as inhibitors of estrogen receptor (ER)-mediated transcription. Bioorg Med Chem 2015;23:4132–8. https://doi.org/10.1016/j.bmc.2015.06.067.Search in Google ScholarPubMed
39. Mao, C, Patterson, NM, Cherian, MT, Aninye, IO, Zhang, C, Montoya, JB, et al.. A new small molecule inhibitor of estrogen receptor α binding to estrogen response elements blocks estrogen-dependent growth of cancer cells. J Biol Chem 2008;283:12819–30. https://doi.org/10.1074/jbc.m709936200.Search in Google Scholar
Supplementary Material
This article contains supplementary material (https://doi.org/10.1515/tjb-2024-0123).
Received: 2024-05-23
Accepted: 2024-06-10
Published Online: 2024-07-18
© 2024 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution 4.0 International License.
留言